Meet our heroes
These people are living with a rare disease and educating the wider public.
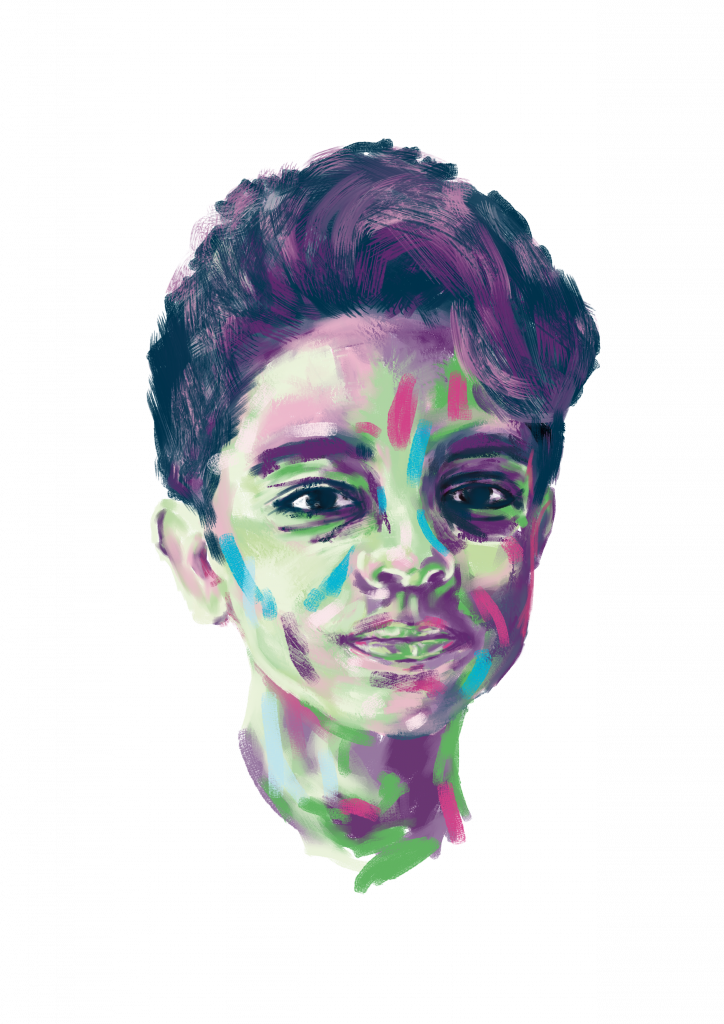
Reza’s Story
Cystinosis
Thirteen-year-old Reza lives in Iran with his family, and he is also living with a rare disease called Cystinosis. Within the past century, Cystinosis has… Continue reading Reza’s Story
Read full story
Nitzia’s Story
Turner syndrome
As a child, Nitzia attended medical appointments for nine years across four different hospitals before she was finally diagnosed with Turner syndrome.
Read full story
Jelena’s Story
Cystic Fibrosis
Jelena finds it difficult to breathe as her lungs do not function properly. Unlike someone who has a more common condition, such as asthma, Jelena… Continue reading Jelena’s Story
Read full story
Shambhavi’s Story
Alagille syndrome
Frequent visits to doctors and hospitals form many of Shambhvai’s earliest memories. To her frustration, she kept being treated for individual symptoms as opposed to… Continue reading Shambhavi’s Story
Read full story
Zixuan’s Story
Mucopolysaccharidosis type 1
Zixuan was two years old when her family started the long process of trying to find a medical diagnosis for her increasing array of symptoms.… Continue reading Zixuan’s Story
Read full story
Tshepiso Gloria’s Story
Von Willebrand’s Disease
For over twenty years Tshepiso Gloria coped with a bleeding disorder before eventually being diagnosed. She went from doctor to doctor, hospital to hospital and… Continue reading Tshepiso Gloria’s Story
Read full story
Wafic’s Story
Duchenne Muscular Dystrophy
Soon after Wafic from Lebanon was born, his parents realised something did not seem quite right. After having spent a worrying few months in hospital… Continue reading Wafic’s Story
Read full story
Syafiq’s Story
Hypohidrotic Ectodermal Dysplasia (HED)
I am Syafiq from Malaysia and I am living with Hypohidrotic Ectodermal Dysplasia (HED). I love hiking in the jungle and jogging (which previously I… Continue reading Syafiq’s Story
Read full story
Regina’s Story
Leiomyosarcoma
Sou mãe de 3 meninos, 2 deles com uma doença rara chamada MUCOPOLISSACARIDOSE Tipo VI. Meu mais velho, Nilton faleceu aos 6 anos devido à… Continue reading Regina’s Story
Read full story
Vasco’s Story
Gaucher Disease
6-year-old Vasco from Peru loves to play, paint, dance and read. But most of all, he loves spending time with his family. It took three… Continue reading Vasco’s Story
Read full story
Nada’s Story
Epidermolysis Bullosa
22-year-old Nada, whose life has been impacted by a rare disease that makes it difficult for her to move, read and swallow, has got a… Continue reading Nada’s Story
Read full story
Taka’s Story
Retinitis Pigmentosa
Hi! My name is Taka, and I’m 36 years old from Japan. I was diagnosed with Retinitis Pigmentosa, a rare eye disease, when I was… Continue reading Taka’s Story
Read full story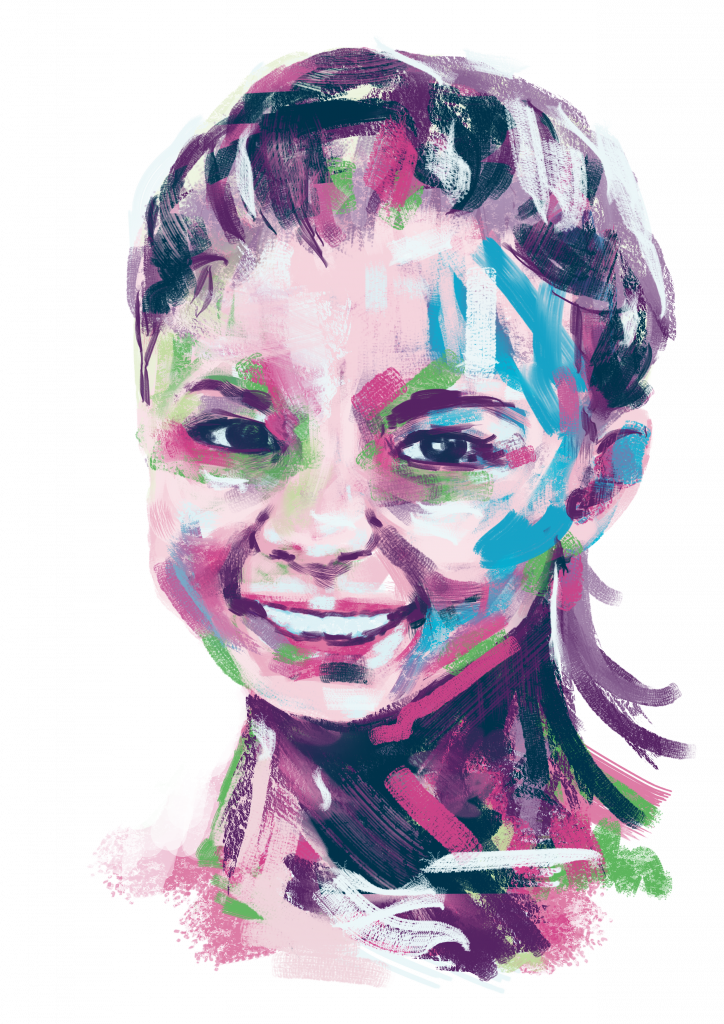
Angelina’s story
Calcium/calmodulin dependent serine protein kinase (CASK)
My 5-year-old daughter, Angelina, is living with CASK-gene related disorders – more precisely, she is living with an X-linked intellectual disability, microcephaly with pontine and… Continue reading Angelina’s story
Read full story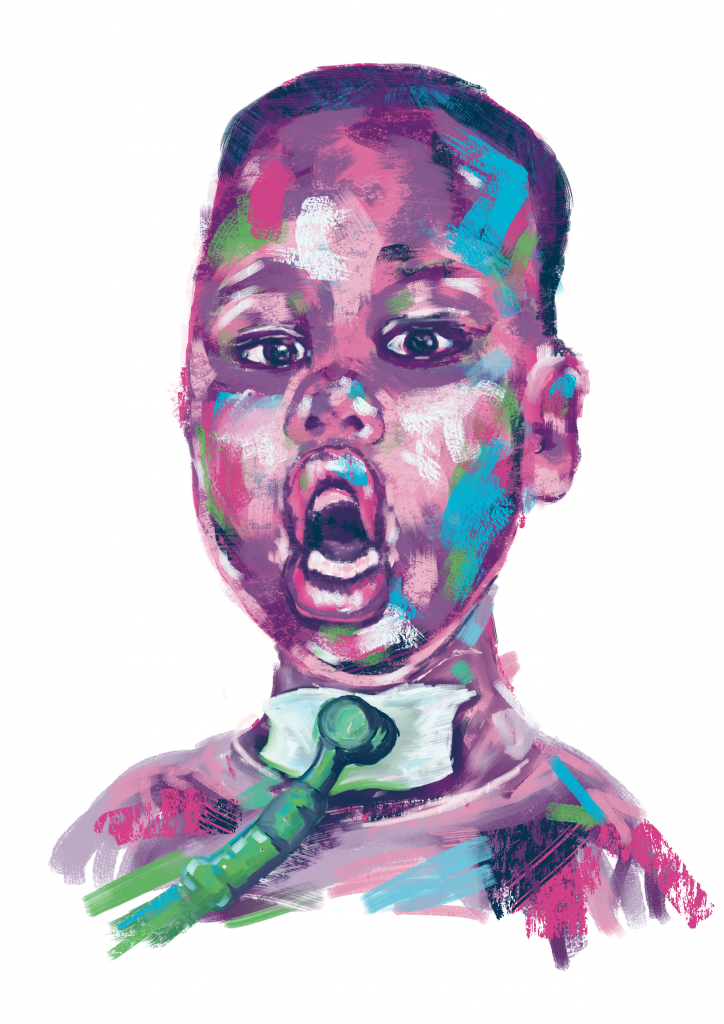
Harvey’s story
Spinal Muscular Atrophy
My son Harvey has a rare condition; SMA, in full Spinal muscular atrophy type 1, which is the more severe type of the disease. Harvey’s… Continue reading Harvey’s story
Read full story
JK’s story
Osteogenesis Imperfecta
My name is Jon-Kristian, but you can call me JK! I am 12-years-old and I live in Norway with my family. I am living with… Continue reading JK’s story
Read full story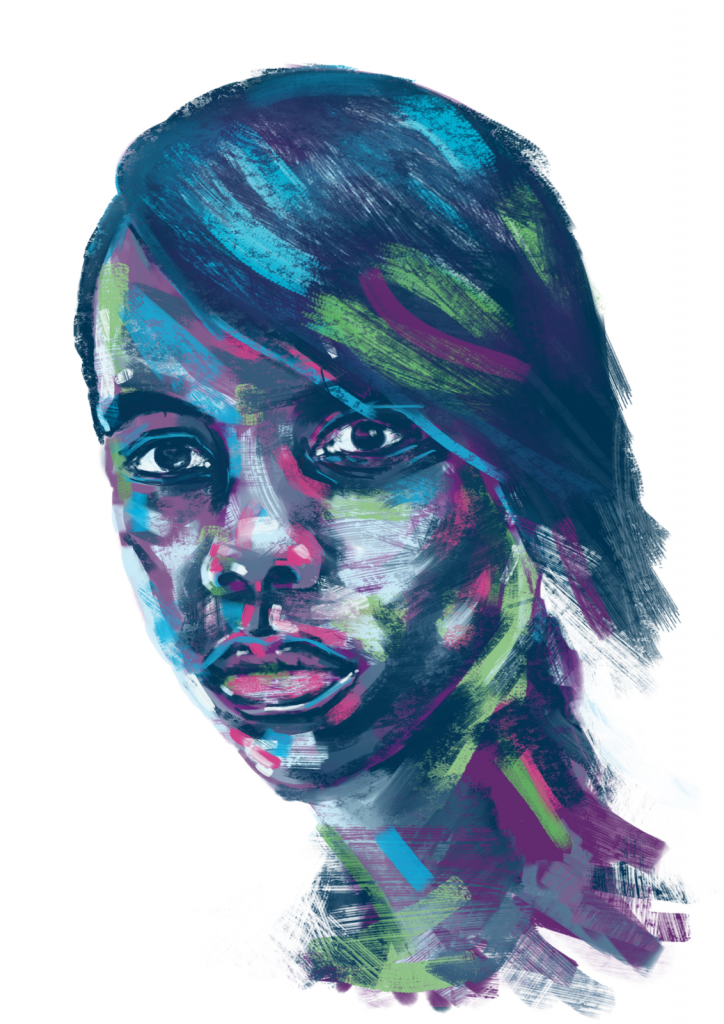
Tristan’s story
Sickle cell anemia
Hello everyone! My name is Tristan from the United States and I am living with Sickle cell anemia. My passion in life is fashion, design,… Continue reading Tristan’s story
Read full story














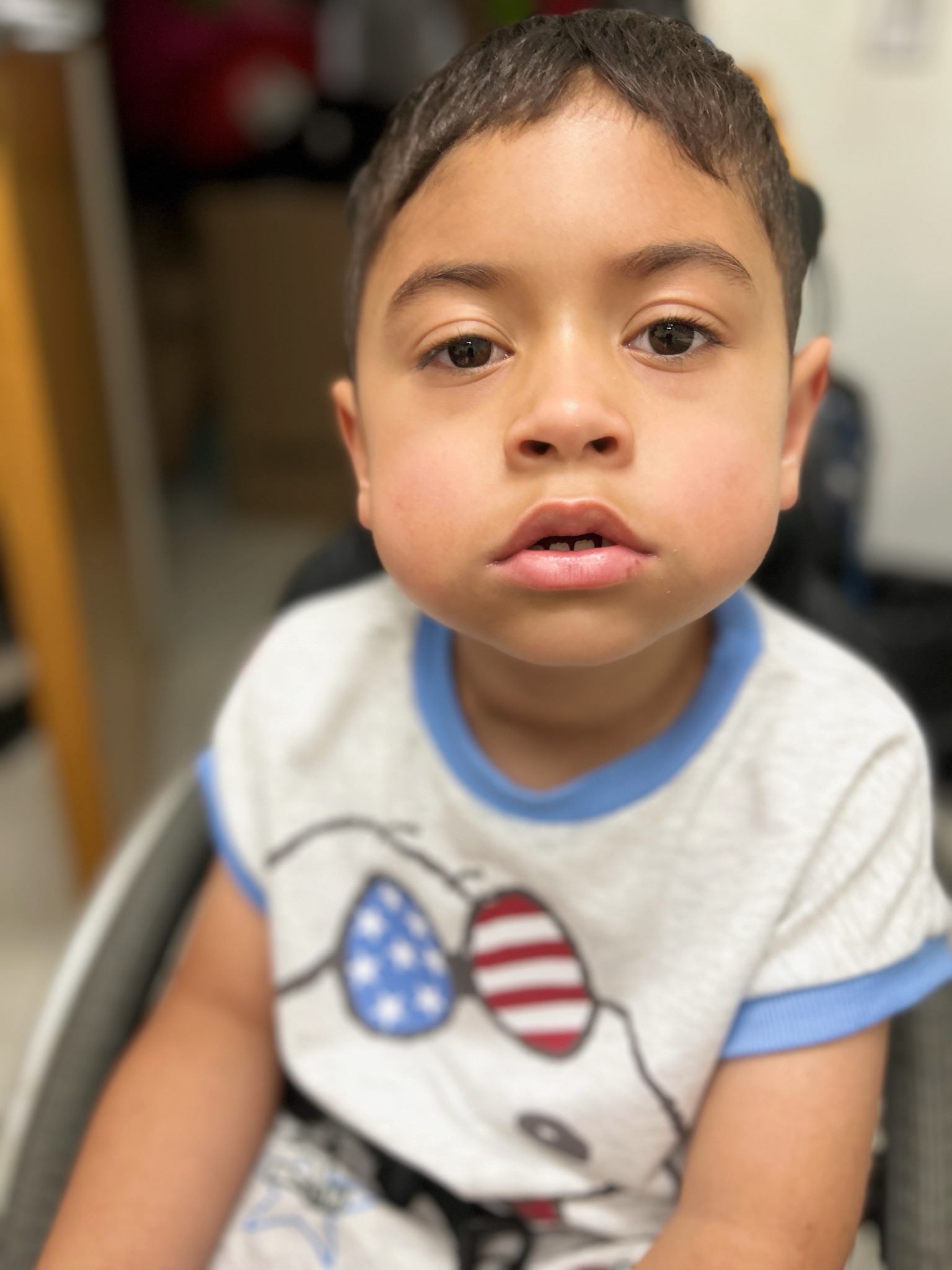

Vincent’s Story
In 2016 my son Vincent was born. Weeks after he was born he started experiencing symptoms of sweating, shaking, paleness and blueness in the lips… Continue reading Vincent’s Story
Read full story
Friends that Care are Rare
“Not Too Rare to Care” Did you know that millions of people are diagnosed with rare diseases and unknown disorders everyday, especially women? According to… Continue reading Friends that Care are Rare
Read full story
Living with Batten Disease: Anthony’s Story
Meet my son Anthony, he was diagnosed with Neuronal Ceroid Lipofuscinosis Type 7, also known as Batten Disease. Batten Disease is a rare, terminal genetic… Continue reading Living with Batten Disease: Anthony’s Story
Read full story
One in a Million
My name is Anna, I am 17 years old, and I am considered ‘one in a million.’ 10 years ago, I was diagnosed with Chronic… Continue reading One in a Million
Read full story
Reaprendendo a viver com Pompe
Sou Magda, 56 anos e tenho Doença de Pompe, uma doença rara, genética e hereditária que causa fraqueza muscular e dificuldade respiratória e apesar de… Continue reading Reaprendendo a viver com Pompe
Read full story
Facciolita and Primary Lymphedema
My Name is Nicole (or facciolita as my social media persona), and I was born with a rare genetic disorder called WILD syndrome that causes… Continue reading Facciolita and Primary Lymphedema
Read full story
Bringing awareness to #rarediseaseday, here is my son’s storytelling…
My name is Baptiste; I am 2 years old. I can already speak very well for my age, I am interested in many things including… Continue reading Bringing awareness to #rarediseaseday, here is my son’s storytelling…
Read full story
Famille sans sucre
Nous sommes une famille porteuse du gène CSID (DCSI). En effet j’ai 35 ans et en 2014 alors que je suis devenue maman pour la… Continue reading Famille sans sucre
Read full story
Léon
Notre fils Léon est atteint de la dystrophie musculaire congénitale type LAMA2, une maladie rare et orpheline d’origine génétique. Lorsqu’il a été diagnostiqué, les médecins… Continue reading Léon
Read full story
MANZI Short Bio
Every parent’s wish is to have a healthy child. Sadly, for us, this wasn’t the case. Eighteen years ago, eight months after giving birth to… Continue reading MANZI Short Bio
Read full story
Carina’s miracle journey
I’m a self taught disabled award winning artist and now an inspirational artist. I was born with a rare genetic disorder called Elhers Danlos Syndrome… Continue reading Carina’s miracle journey
Read full story
Aaron Loving Determination
During the summer of 2021, Aaron started to complain about his hearing and unable to see. This led us down a rabbit hole of doctors… Continue reading Aaron Loving Determination
Read full story
A vida é uma dádiva, viva e agradeça por estar vivo!
Sou Mariana (37 anos), de Brasil. Nasci sem complicações no parto, mas foi observado a falta de pêlos no corpo todo (os cabelos da cabeça… Continue reading A vida é uma dádiva, viva e agradeça por estar vivo!
Read full story
Wings of Fire: escaping the shadows of a rare disease that was undiagnosed for 25 years
Since childhood, I have always looked physiologically thin. When I turned 4, my parents recognised that I was a little different compared to other kids… Continue reading Wings of Fire: escaping the shadows of a rare disease that was undiagnosed for 25 years
Read full story
God’s Grace
During my 20 week anatomy scan we learned that our baby had arthrogryposis. The doctors told us she could not and would not ever move… Continue reading God’s Grace
Read full story
My motherhood battle with a ghost disease
Im sure you laughed when you read the title but yeah I call it that because just like a ghost my this disease keeps appearing… Continue reading My motherhood battle with a ghost disease
Read full story
Andryusha’s Story
My husband and I had our first son, Andryusha, in 2011. Despite seeming happy and healthy at birth, we consulted with his doctor when he… Continue reading Andryusha’s Story
Read full story
Policondritis Recidivante
Policondritis Recidivante La Policondritis Recidivante (PR) es una rara enfermedad inmunomediada que se caracteriza por la inflamación y en la destrucción del cartílago. Todos los… Continue reading Policondritis Recidivante
Read full story
Haciendo visible lo invisible
Mi nombre es Diego, tengo 14 años de edad y soy de México, mi vida entera la he pasado en hospitales; por medio de estudios… Continue reading Haciendo visible lo invisible
Read full story
Máximo el Grande
Durante el embarazo de Máximo no se sospecho de nada era un embarazo normal, sin complicaciones, todas sus radiografías estaban dentro de parámetros normales excepto… Continue reading Máximo el Grande
Read full story
Everything sucks but I’ am STILL happy
Als Kleinkind konnte niemand herausfinden, warum ich wochenlang hohes Fieber hatte und nicht mehr gelaufen bin; Keiner konnte sich in meiner Jugendzeit meine Gelenkprobleme erklären.… Continue reading Everything sucks but I’ am STILL happy
Read full story
Diagnosis is not Destiny
My name is Celyna, and I am from Natal, Rio Grande do Norte, Brazil. I have been diagnosed with hereditary spastic paraparesis subtype SPG4. My… Continue reading Diagnosis is not Destiny
Read full story
Gideon, The Little Warrior
I could lie and say Gideon started his life as an easy baby, but I will not. Gideon started his life with a BANG. He… Continue reading Gideon, The Little Warrior
Read full story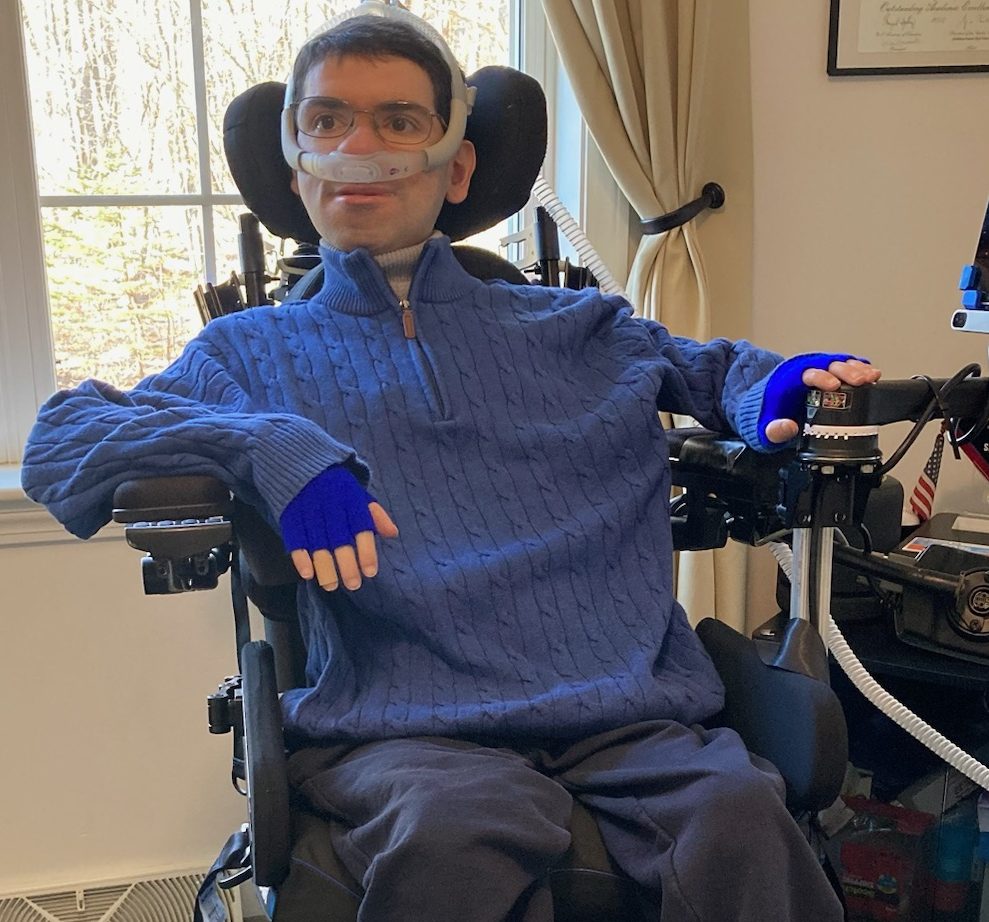
Sachin: Resilience in the face of Duchenne Muscular Dystrophy
My name is Sachin and I’m a 37-year-old guy who has a rare genetic disease known as Duchenne muscular dystrophy (DMD), which primarily affects males,… Continue reading Sachin: Resilience in the face of Duchenne Muscular Dystrophy
Read full story
Seeking a Cure for Simon
When my wife Edith was pregnant with Simon, our first child, we were filled with joy and excitement for what was to come. That joy… Continue reading Seeking a Cure for Simon
Read full story
Eliza’s story
Eliza has two rare conditions, Beckwith Wiedemann Syndrome and Metopic Craniosynostosis. Eliza’s story began when the midwife noticed Eliza had an abnormally large and long… Continue reading Eliza’s story
Read full story
I’m Not A Mystery
I was faced with an unnamed disease that tormented me for seven long years. This sudden and excruciating ailment left me with enduring pain, making… Continue reading I’m Not A Mystery
Read full story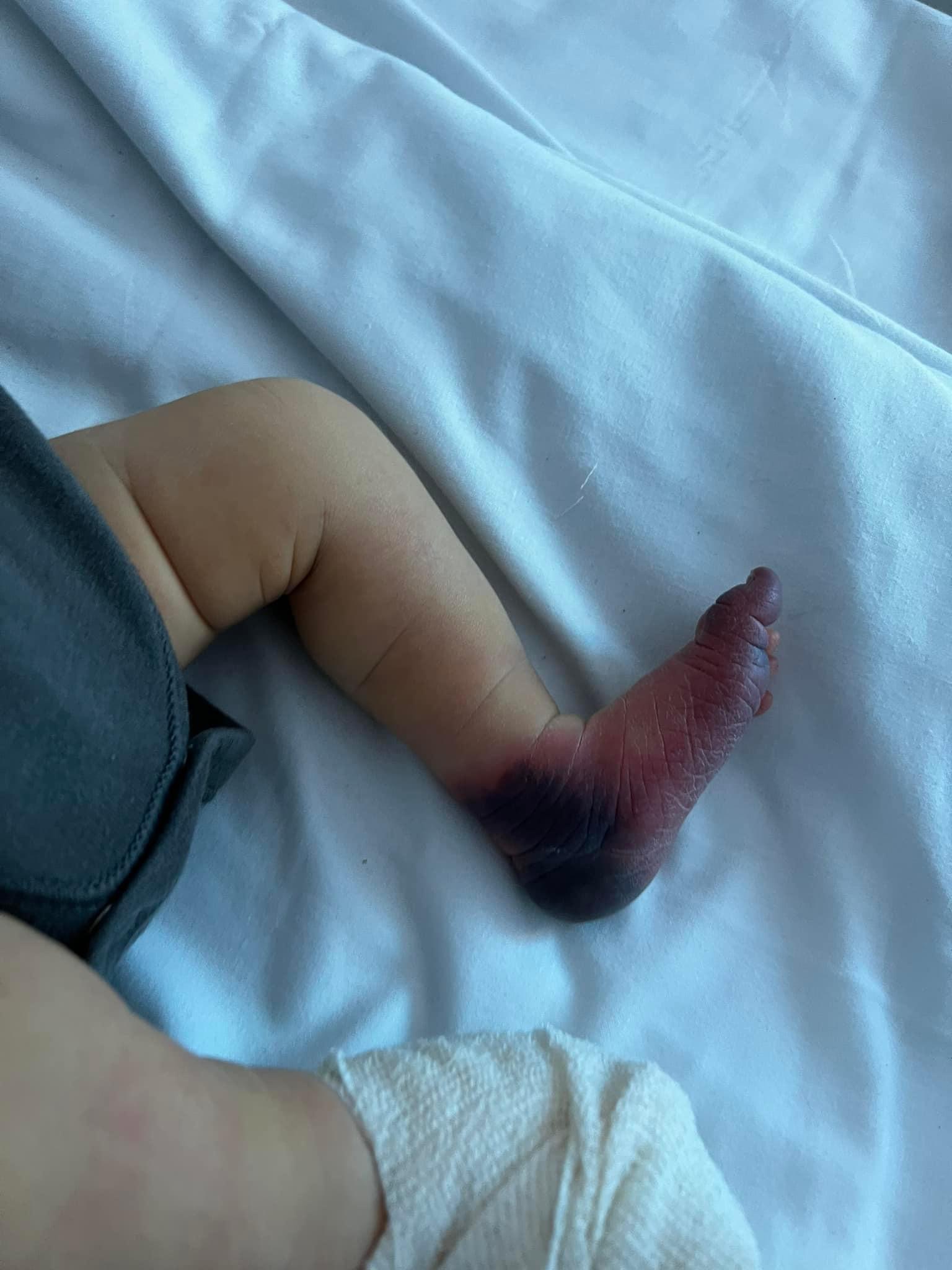
Hector’s triumph
We had our son Hector on the 19th May 2023, he was a seemingly healthy baby and we couldn’t have been more smitten with him.… Continue reading Hector’s triumph
Read full story
Light of life
Vietnamese version right here, English version is below. Xin chào cả thế giới, Tôi là Đỗ Phước Huy, 30 tuổi, và tôi đến từ Việt… Continue reading Light of life
Read full story
MEN-1 might be rare but hope doesn’t have to be
Sharing my husbands story. My husband is a MEN 1 fighter a long with our daughter who is 6. Brian was diagnosed with MEN-1 at… Continue reading MEN-1 might be rare but hope doesn’t have to be
Read full story
My Ataxia journey…
My adventure with this condition began at the age of 16, when I started to lose my balance and got a drunken gait. Over time,… Continue reading My Ataxia journey…
Read full story
West Yorkshire family fight against rare disease
I am based in Skelmanthorpe, with my husband James, and my two children Elijah (4) and Stefan (5 months). I am writing to submit a… Continue reading West Yorkshire family fight against rare disease
Read full story
Living with the Challengers of a Complex Lymphatic Anomaly
Alfie, now 15, is a typical teenager giving me attitude. After receiving the diagnosis of lymphangiomatosis in 2009 I did not think we would ever… Continue reading Living with the Challengers of a Complex Lymphatic Anomaly
Read full story
A Cure for Sophia and Friends
Sophia Rose was born in 2007 to proud parents Rachel and Gary in Laguna Beach, CA, USA. We did not receive the diagnosis of her… Continue reading A Cure for Sophia and Friends
Read full story
Sharing Anthony’s Journey: Navigating the World of Galloway-Mowat Syndrome (as told by his mom!!)
Meet my son Anthony, a courageous 4.5-year-old warrior with a spirit that inspires us all. Anthony’s journey began with a diagnosis at just 2 months… Continue reading Sharing Anthony’s Journey: Navigating the World of Galloway-Mowat Syndrome (as told by his mom!!)
Read full story
“Verily, With Hardship Comes Ease”
My name is Sareena. I was born on September 21, 1995. I was diagnosed with severe Factor VII Deficiency when I was two months old.… Continue reading “Verily, With Hardship Comes Ease”
Read full story
Love for Loredana
Loredana has a smile that lights up your soul, she can’t get enough hugs from her family members, and watching the family pets provides hours… Continue reading Love for Loredana
Read full story
BOR Warrior
I’m Lily and I’m 12. I have a rare condition called Branchiotorenal Syndrome. What does that mean? It means that I am deaf/ hard of… Continue reading BOR Warrior
Read full story
Using a Lifetime of Rare Disease to Change the Future
I’m Jenny and I’m a Rare Disease Advocate for Familial Adenomatous Polyposis (FAP) and Short Bowel Syndrome (SBS). I was diagnosed with FAP at age… Continue reading Using a Lifetime of Rare Disease to Change the Future
Read full story
A cdg journey
I was born as a normal child until one day my mom noticed I was turning blue with my arms up and eyes up. That… Continue reading A cdg journey
Read full story
Mein Leben mit EB
Mein Name ich Clara, ich bin 24 Jahre alt und wurde mit der seltenen Hautkrankheit Epidermolysis bullosa – kurz EB geboren. Meine Haut ist so… Continue reading Mein Leben mit EB
Read full story
Adapting to life with Dystonia
My life became very strange in the Summer of 2001. While pursuing my master’s degree in counseling, I noticed that my head was slightly tilting… Continue reading Adapting to life with Dystonia
Read full story
My experience living with EPP
I am Sam, a 21 year old film student living in the UK. I have recently created a short documentary for my course about living… Continue reading My experience living with EPP
Read full story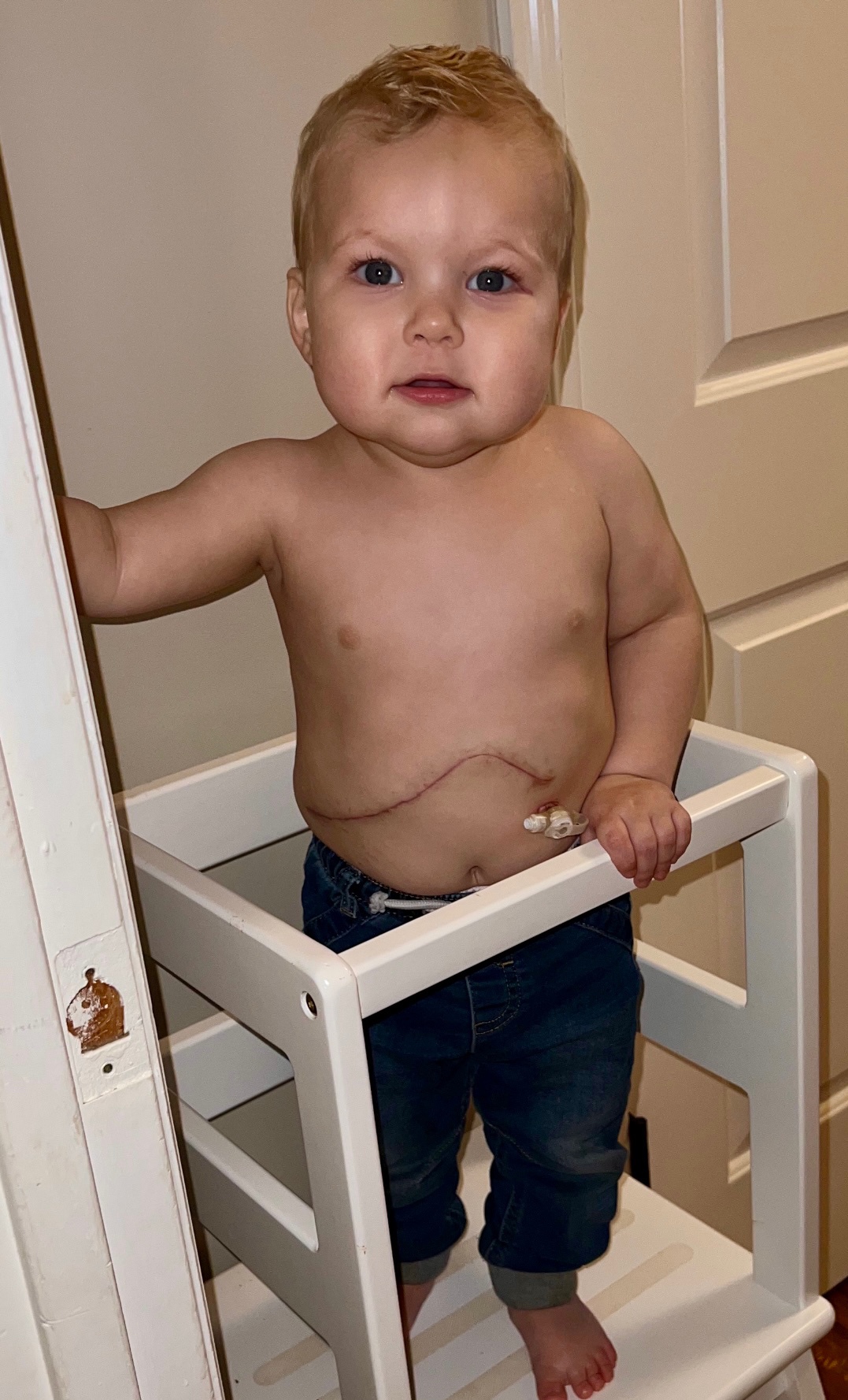
Rare Disease and Liver Transplant Warrior
Our son Cameron was born a seemingly healthy baby on November 4, 2022. After two days in the hospital, we were sent home with him… Continue reading Rare Disease and Liver Transplant Warrior
Read full story
The Brain is a misunderstanding.
My name is Kim and this is my focus story of rare disorder awareness. I know personally what it is — to have a rare… Continue reading The Brain is a misunderstanding.
Read full story
A dor é inevitável! O sofrimento é opcional !
Em 2017, aos 23 anos depois de fazer uma RM crânio cervical, e de inúmeros sintomas veio o diagnóstico que por sua vez tinha um… Continue reading A dor é inevitável! O sofrimento é opcional !
Read full story
Fighting with fragile bone
300 Million people with rare diseases 600+ events worldwide 106+ countries involved When my son Animesh was diagnosed with Osteogenesis Imperfecta (OI), doctors told me… Continue reading Fighting with fragile bone
Read full story
Fabio und sein Plexusarm
Heute spreche ich für meinen Sohn Fabio, der am 18.09.23 mit einer geburtstraumatischen Plexusparese geboren ist. Eine Plexusparese ist eine Armlähmung durch eine Verletzung der… Continue reading Fabio und sein Plexusarm
Read full story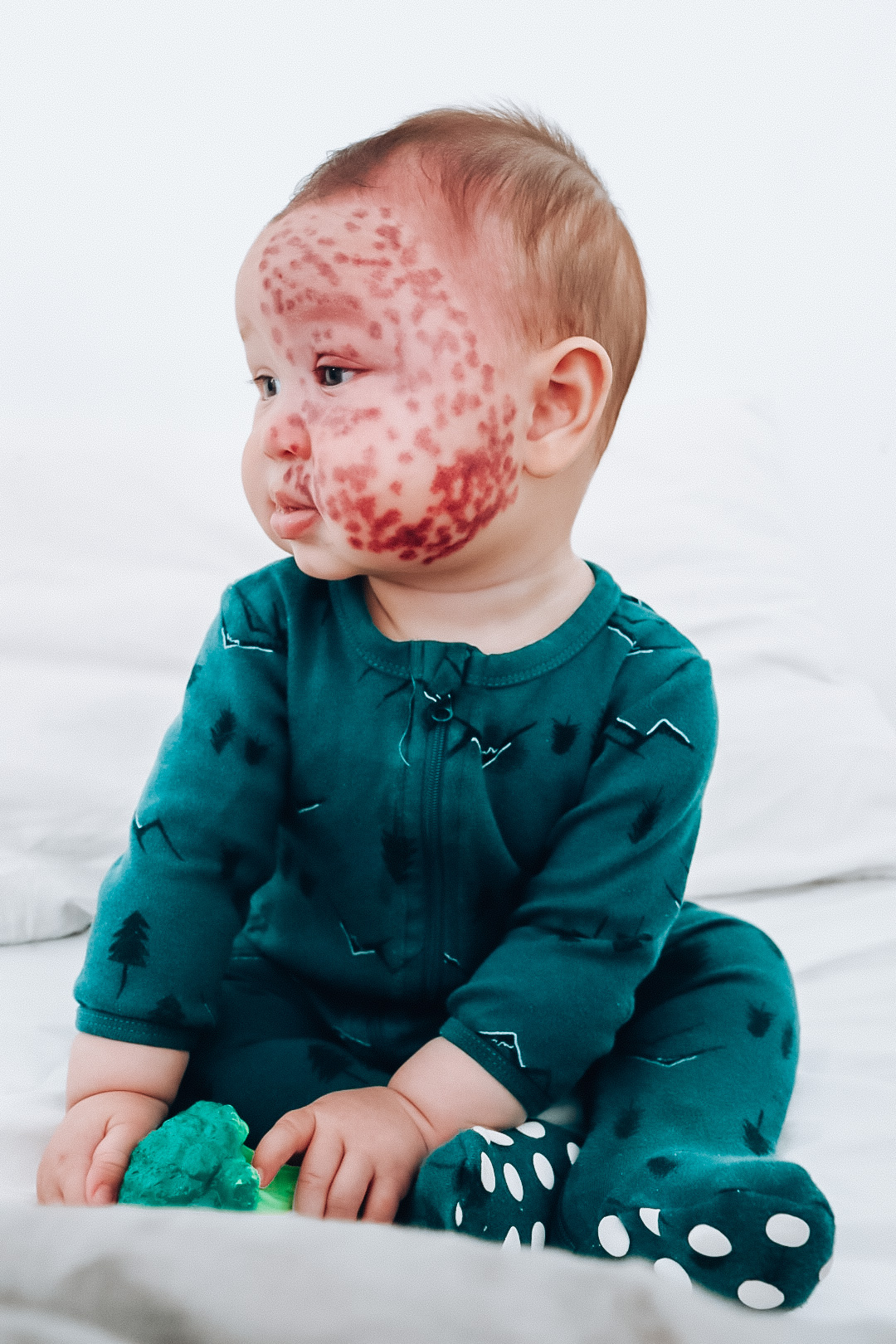
A Sturge-Weber mini warrior!
My 2 year old little man Kingsley, is the definition of a Sturge-Weber warrior! Born with a large Port-wine Stain Birthmark across half of his… Continue reading A Sturge-Weber mini warrior!
Read full story
One in a Million can be quite lonely
According to Google, the furry, adorable koala sleeps anywhere between 20-22 hours a day for survival, making it the sleepiest animal in the world. Apparently,… Continue reading One in a Million can be quite lonely
Read full story
I am still Katie
My name is Katie and I am from Spokane, Washington. Currently, I am in school pursing a career in nursing. I have Hereditary Angioedema. I… Continue reading I am still Katie
Read full story
Povestea mea cu Ataxia Spinocerebeloasa…
Mă numesc Mitre Anamaria din Zalău, jud. Sălaj, am 41 de ani şi de 24 de ani trăiesc cu o boală rară. Diagnosticul meu este… Continue reading Povestea mea cu Ataxia Spinocerebeloasa…
Read full story
Long jurney
Hello. My name is Agata. Im 41 years old. Im from Poland but live in Scotland for 14 years. Everything started 2011 on February with… Continue reading Long jurney
Read full story
Transformando corazones en #iFopers. Única en 2 millones
Tengo Fibrodisplacia Osificante Progresiva (FOP). Cuando yo nací, tenía una formación distinta en ambos pies, los médicos nos decían que tal vez a mi me… Continue reading Transformando corazones en #iFopers. Única en 2 millones
Read full story
My life with HNPP
As a nurse working in the height of COVID in 2020, while raising 2 little children ages 5 and 2, I began having strange symptoms.… Continue reading My life with HNPP
Read full story
From Teacher to Moyamoya Warrior
My name is Melissa and I reside in Cleveland, Ohio, with my amazing husband, Luke and our dog, Duke. I have always loved kids and… Continue reading From Teacher to Moyamoya Warrior
Read full story
d’une suspicion de déshydratation à un chylothorax bilatéral réfractaire
Je m’appelle Aleeza, j’avais 4.5 ans quand j’ai été prise de vomissements … ma maman qui n’est pas d’une nature stressée a tout de même… Continue reading d’une suspicion de déshydratation à un chylothorax bilatéral réfractaire
Read full story
My Journey with Wilsons Disease
My name is Nicolette. I am 21 years old and was diagnosed with Wilsons Disease in October 2023. Wilsons disease has not been known to… Continue reading My Journey with Wilsons Disease
Read full story
Shining Through Darkness: Sara’s Journey with BPAN, a Tale of Strength and Hope
Sara, a remarkable young lady who embodies the spirit of courage and resilience in the face of a rare neurodegenerative disorder known as Beta-Propeller Protein-Associated… Continue reading Shining Through Darkness: Sara’s Journey with BPAN, a Tale of Strength and Hope
Read full story
The Stranger that lives between my Bones
She did not have a name for the longest time; all I knew was that she brought a pain I never knew existed. She would… Continue reading The Stranger that lives between my Bones
Read full story
How A Rare Neuromuscular Disease Saved My Life
Strange Symptoms My jaw literally dropped. Chewing, speaking, and swallowing led to severe bulbar exhaustion, and the inability to move my jaw. The symptoms progressed… Continue reading How A Rare Neuromuscular Disease Saved My Life
Read full story
Celebrating Life and Spreading Awareness in Cancer Survivorship
When I was 21 years old, I found out I was pregnant. I didn’t know that the pregnancy would be the only surprise that year,… Continue reading Celebrating Life and Spreading Awareness in Cancer Survivorship
Read full story
Turning Pain Into Purpose With Rare Disease Girl
After giving birth to my baby, I triggered an idiopathic rare disease called Atypical Hemolytic Uremic Syndrome. It caused a cascade of life-threatening issues including… Continue reading Turning Pain Into Purpose With Rare Disease Girl
Read full story
My journey is one of resilience, self-advocacy, and hope.
My name is Rain, From my earliest memories, I have carried the weight of excruciating stomach pains. These pains stole away the joys of a… Continue reading My journey is one of resilience, self-advocacy, and hope.
Read full story
Turning grief into purpose
I’m Carla, a mom of a Hirschsprung’s Disease (HD) child -who passed away at the age of 14 months- due to an undiagnosed enterocolitis. For… Continue reading Turning grief into purpose
Read full story
Tommy’s RTD Journey (as told by his mom!)
Before his diagnosis, Tommy was a normal, happy little boy. He was a little developmentally delayed, but we didn’t think much of it. He was… Continue reading Tommy’s RTD Journey (as told by his mom!)
Read full story
My story with Anorrectal Malformation
Primeramente les cuento que vivimos en Tandil, Provincia de Buenos Aires. En el 2017 nace mi segundo hijo Fermín. Era muy extraño todo con él,… Continue reading My story with Anorrectal Malformation
Read full story
Love Isn’t Rare
Growing up in a family impacted by Huntington’s Disease – a rare and incurable genetic disorder, I feel compelled to share my exceptional life experiences… Continue reading Love Isn’t Rare
Read full story
Benjamin’s journey
Benjamin was about 3 months old when he had his first seizure (that we know of), he was sent to sick kids hospital in Toronto… Continue reading Benjamin’s journey
Read full story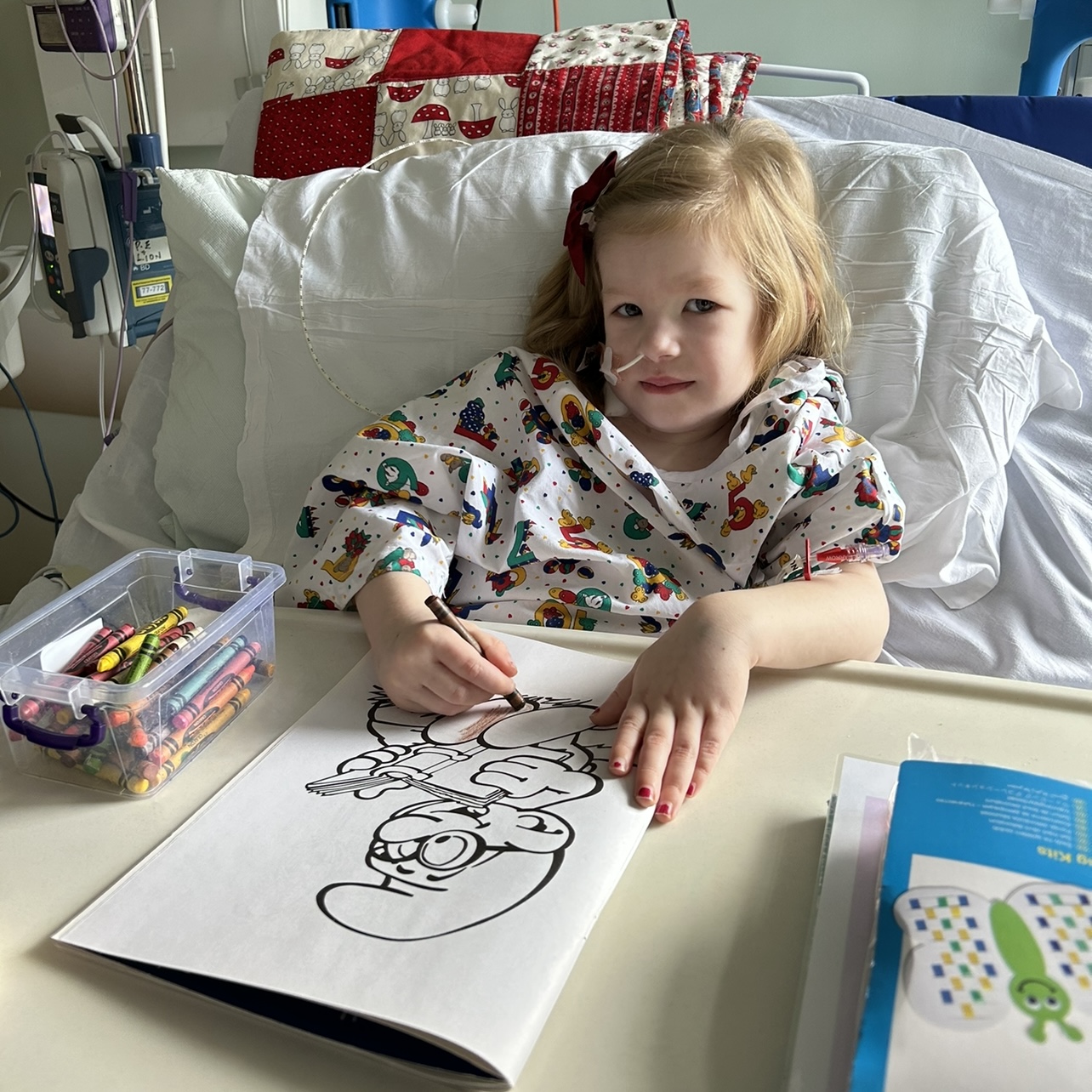
Evie’s story
Evie was diagnosed with a very rare life threatening genetic condition called LPIN1 in April 2023 age 4 years old. Caused by both parents having… Continue reading Evie’s story
Read full story
Yeni’s Story
Hi, my name is Yeni, from Indonesia. I have rare disease called Friedreich’s Ataxia. I’m diagnosed with Friedreich’s Ataxia (FA) when I was 19 years… Continue reading Yeni’s Story
Read full story
Mighty Morgan
Morgan’s journey began innocently enough, with her parents, Darius and Laura, eagerly embracing the joys of parenthood. However, after coming home from the hospital, they… Continue reading Mighty Morgan
Read full story
My baby broke my back
I first felt pain in my back in July 2019, when my first baby was a month old. It just got worse and worse, and… Continue reading My baby broke my back
Read full story
Two of a kind
Jaxon (age 14) and Jeren (age 10) are brothers who mirror each other with genetics. Both boys have multiple gene deletions that have resulted in… Continue reading Two of a kind
Read full story
Amelia, CLN1 Batten Disease Warrior!
Our daughter, Amelia, was diagnosed with CLN1 Batten Disease (neuronal ceroid lipofuscinosis) at 2 years old. Before learning of her diagnosis, we had never heard… Continue reading Amelia, CLN1 Batten Disease Warrior!
Read full story
Non sono quello che mi è successo. Sono quello che ho scelto di essere. Carl Gustav Jung
Hai presente quando alla fine di un film giallo svelano il nome dell’assassino e tutti gli indizi sparsi ore precedenti trovano finalmente una collocazione? Ecco,… Continue reading Non sono quello che mi è successo. Sono quello che ho scelto di essere. Carl Gustav Jung
Read full story
1 heure à la fois
À l’occasion de la Journée des Maladies Rares, je souhaite partager avec vous mon parcours avec le Syndrome de Schmidt. Depuis septembre 2021, je vis… Continue reading 1 heure à la fois
Read full story
Katie’s Story
My name is Katie and I was diagnosed with CMTC when I was 5 months old. I am now 30 years old and have lived… Continue reading Katie’s Story
Read full story
Annarita e la sua passione per la danza oltre la disabilità
Mi chiamo Annarita ho 27 anni e sono residente a Pagani, un paese in provincia di Salerno. Il destino ha voluto che proprio il giorno… Continue reading Annarita e la sua passione per la danza oltre la disabilità
Read full story
Melodies of Resilience: Chapman’s Undiagnosis Disease and Musical Triumph
Chapman, an extraordinary 12-year-old blind pianist, has embarked on an incredible journey since his birth. Born in Hong Kong and currently residing in London, Chapman… Continue reading Melodies of Resilience: Chapman’s Undiagnosis Disease and Musical Triumph
Read full story
Brittle but not broken
At three and a half months old, my first fracture appeared. Within a year, I had fractured three more bones. The doctor after having a… Continue reading Brittle but not broken
Read full story
Josiah’s Journey through FCAS
Josiah is 23 months old and been having symptoms since birth! Starting with 3 infections at 10 days old, which lead to a spinal tap… Continue reading Josiah’s Journey through FCAS
Read full story
My Mast Cell Disease Story
In 2018 I was diagnosed with CIRS: Chronic inflammatory response syndrome, Adrenal Insufficiency, and was also believed to be Mast Cell Disease. I had a… Continue reading My Mast Cell Disease Story
Read full story
Leo’s story
Our little Leo was born at 37 weeks after an emergency scan He was released from the hospital two days after birth with no health… Continue reading Leo’s story
Read full story
Oakley’s Journey
Oakley was diagnosed with Nicolaides-Baraitser Syndrome in October 2019. When I was pregnant with Oakley, we knew we wanted to choose a good, strong name… Continue reading Oakley’s Journey
Read full story
A little insight to my daily life!
My name is Amber and I live in Queensland. I was born with Netherton Syndrome which is a form of Ichthyosis. Ichthyosis is an extremely… Continue reading A little insight to my daily life!
Read full story
Vivir en un mundo de 5 dedos
Hola soy Ana Victoria una niña de 8 años que a vivido en un mundo de personas con 5 dedos en cada mano, siempre busco… Continue reading Vivir en un mundo de 5 dedos
Read full story
Nevidljiva
Lijep pozdrav. Moje ime je Senaida, a moj sin se zove Dželal, koji boluje od rijetkog oboljenja, cistične fibroze. Živimo u Srebreniku mali gradić u… Continue reading Nevidljiva
Read full story
Je transcende mon handicap
Je m’appelle Julien, j’ai 34 ans et je suis atteint d’une Myopathie de Duchenne. Il s’agit d’une maladie génétique rare qui provoque une dégénérescence musculaire… Continue reading Je transcende mon handicap
Read full story
Синдром на Гителман
Здравейте казвам се Камелия, медицинска сестра съм от България.Синът ми се казва Максим и страда от рядко генетично заболяване Синдром на Гителман.Още от много малък… Continue reading Синдром на Гителман
Read full story
Short leg but hopeful eyes
Sofia is 1 year and 9 months old. She has congenital defect. Her left leg is shorter than right leg by 9,5 cm. And until… Continue reading Short leg but hopeful eyes
Read full story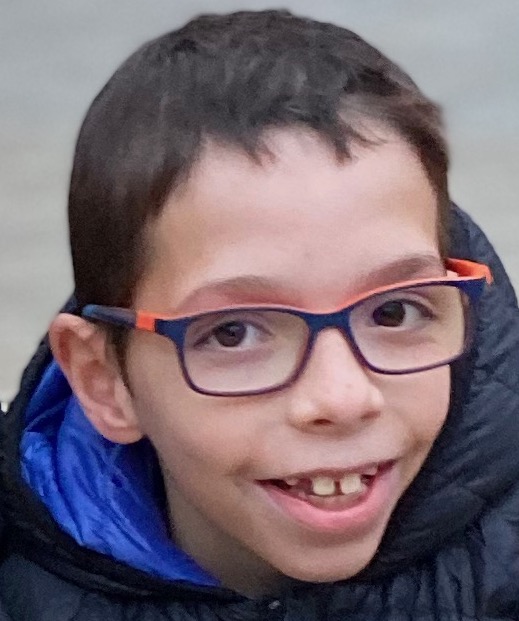
MARC, A HAPPY FACE DESPITE HIS RPS6KA3 GENE MUTATION
Marc, our son, is a wonderful 11-year-old enjoying life in the green, residential town of Sant Cugat (Barcelona, Spain). Not only does he fill our… Continue reading MARC, A HAPPY FACE DESPITE HIS RPS6KA3 GENE MUTATION
Read full story
Ліза із Маріуполя .
Доброго дня . Чудова , життєрадісна Ліза , із міста Марії, із Маріуполя . Нам пощастило вибратися із окупації , та втекти від війни .… Continue reading Ліза із Маріуполя .
Read full story
Sentirsi diversi senza esserlo
Sono Valentina, ho 23 anni, appena arrivata in Italia a 7 anni mi hanno diagnosticato a Udine hypoglycemia una malattia rara che pochissimi conoscono. La… Continue reading Sentirsi diversi senza esserlo
Read full story
Living with CDH
I was 27 weeks pregnant when my MFM confirmed our son would be born with congenital diaphragmatic hernia. He had right sided CDH, with liver… Continue reading Living with CDH
Read full story
Not Too Rare To Care
Hi, my name is Jayme and I am 26 years old. When I was born in 1996, my Mom along with Doctors knew there was… Continue reading Not Too Rare To Care
Read full story
My Story My Journey (قصتي رحلتي)
Hello ? My name is Omaima but you can call me om , I’m from Kuwait city ??? Have arteriovenous malformation (avm ) left leg… Continue reading My Story My Journey (قصتي رحلتي)
Read full storyWhat's your story?
Be part of Rare Disease Day by sharing your story with others and sending a message of solidarity!
Share your storyShare your colours
Join the community. Help us build awareness. Share your photos, videos and experiences!
Meet our heroes
Read their stories
Become our friend
Sign up your organisation
Find an event near you
Join the campaign
Download materials
Share your colours